中国科学院高能物理研究所蔡肖夏课题组与北京大学、华盛顿大学等机构组成的研究团队,提出了一种能够精确构建分子势能面并实现混合量子–经典非绝热动力学演化的量子算法架构。相关成果以“Efficient Quantum Simulation of Non-Adiabatic Molecular Dynamics with Precise Electronic Structure”为题,于2026年1月正式发表于《数字发现》(Digital Discovery 5, 548 (2026)),并被选为封面文章。
非绝热分子动力学是研究光催化、光合作用等关键化学与生物过程的重要理论工具,其核心挑战在于精确描述多个电子态结构,以及分子体系在这些电子态上的精确含时演化。传统计算方法在处理高维电子结构与非绝热动力学时面临指数墙问题。尽管量子计算在电子结构模拟方面具有重要的潜力,但如何将量子算法高效、精确地嵌入非绝热动力学框架,仍是制约其实际应用的关键难题。
针对上述挑战,研究团队提出了一种量子计算适配的扩展Landau–Zener面跳跃(LZSH)非绝热分子动力学框架。通过引入曲率保护的跳跃修正,在避免计算非绝热耦合(NAC)的情况下,保持了电子态布居数演化的物理一致性,从而能够有效地描述电子态的跃迁过程。此外,为满足LZSH动力学对高精度势能面的需求,该工作开发了一种高精度(误差在1*10-6 Hartree量级)的势能面计算方案。该方案支持活性空间选择,可在量子或经典计算集群上实现并行加速,并展示了对多种化学体系(如H3+离子、乙烯分子等)的良好适应性。
该研究为量子计算在高精度化学动力学模拟领域提供了重要参考。工作所构建的高精度势能面计算与非绝热分子动力学框架,为精确刻画激发态能量转移、质子转移等光物理与光化学过程开辟了新路径,也为复杂分子体系的精细化模拟奠定了坚实基础。
该工作的第一作者为北京大学前沿计算科学研究中心的李天翊,通信作者为北京大学的袁骁、中国科学院高能物理研究所的蔡肖夏以及华盛顿大学的汤典东。本研究得到了国家自然科学基金等项目的支持。
论文链接:https://pubs.rsc.org/en/content/articlelanding/2026/dd/d5dd00433k

期刊封面
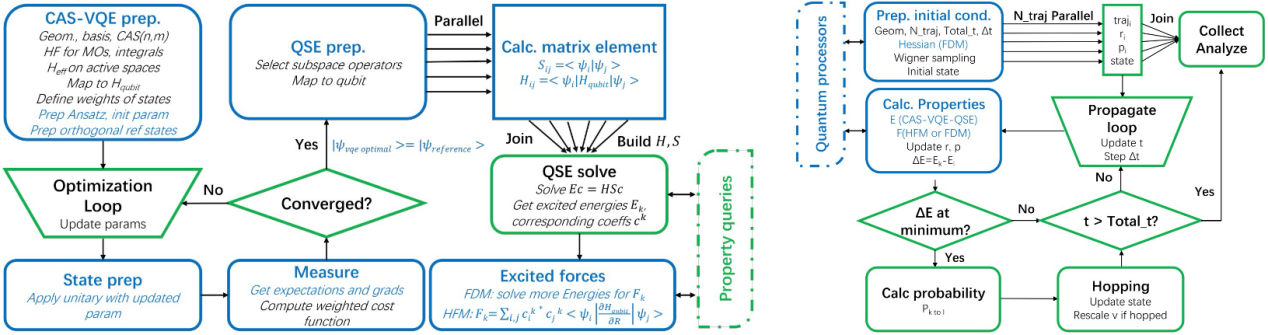
混合量子-经典非绝热动力学算法架构:左图为电子结构计算流程图;右图为扩展LZSH非绝热分子动力学流程。
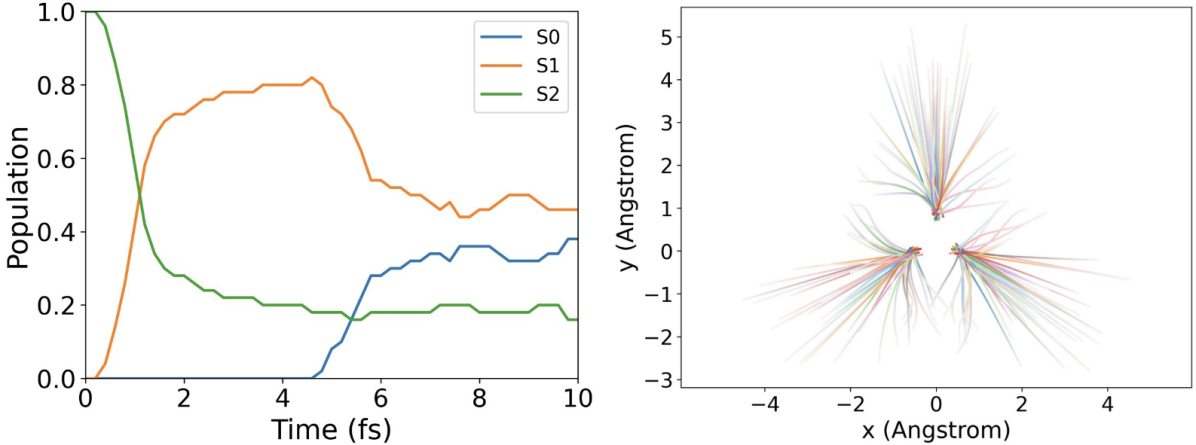
H3+的模拟结果:左图为各电子态布局数随时间的变化;右图为氢核的模拟轨迹。
附件下载: